
邮 箱:info@tlph.com.la
在7000多种已知的罕见病中,肺动脉高压患者在全球就有超过2500万人,是非常不罕见的“罕见病”。主要因为遗传基因突变和心脏疾病相关造成的肺动脉压力升高导致身体长期处于缺氧状态,让“呼吸”这件再普通不过的事,在肺动脉高压患者中变成了一场艰难的战役。
根据世界卫生组织(WHO)的分类,肺动脉高压(PH)分为五类:第一类是动脉型,第二类左心疾病相关,第三类肺部疾病或低氧相关,第四类慢性血栓栓塞性,第五类不明原因或多因素。肺动脉高压因其发病机制隐匿容易被误诊,同时具有高致死率和并发症的风险,对生命造成重大威胁,需要社会更多关注。
目前东盟制药(TLPH)生产的马昔替坦和他达拉非获得老挝卫生部食品药品(FDD)已经获批上市!
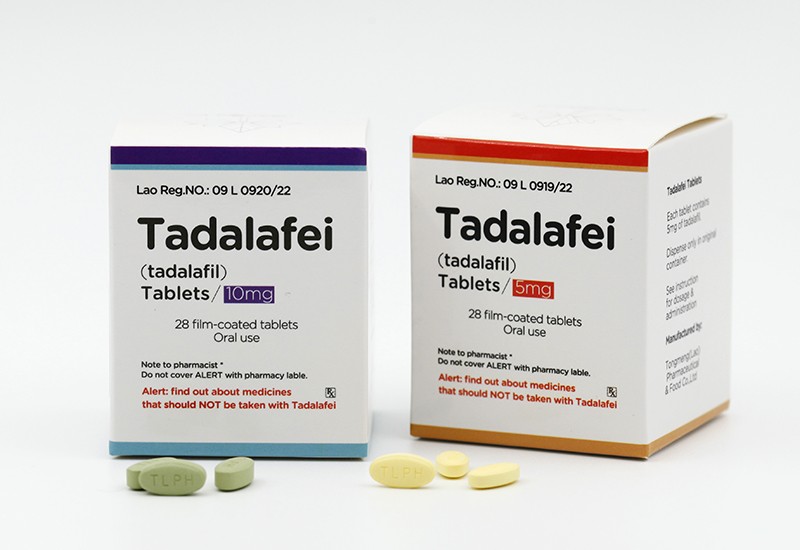
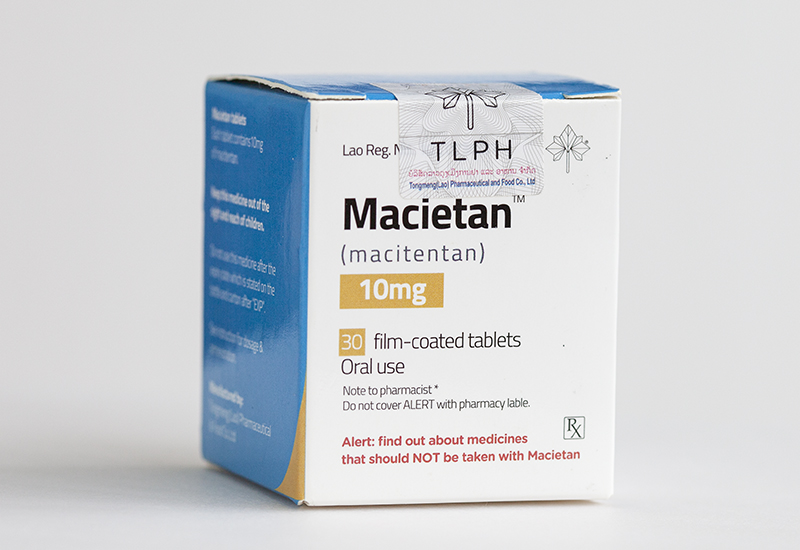
以下是macitentan和tadalafil治疗PAH的作用机制:
macitentan:作为双重内皮素受体拮抗剂(ERA),通过阻断内皮素-1(ET-1)与ETA和ETB受体结合,抑制ET-1介导的血管收缩、平滑肌细胞增殖及纤维化等病理过程,从而降低肺血管阻力,延缓疾病进展124。其对肺动脉平滑肌细胞具有高亲和力,且代谢产物仍保留部分药理活性13。
tadalafil:作为磷酸二酯酶5(PDE5)抑制剂,通过抑制PDE5酶活性,减少环磷酸鸟苷(cGMP)的降解,从而增强一氧化氮(NO)介导的血管舒张作用,改善肺动脉血流动力学并降低肺动脉压力6711。
PAH的危害与治疗迫切性:
肺动脉高压(PAH)是一种罕见且致命的血管疾病,以肺小动脉狭窄、肺循环压力升高为特征,最终导致右心衰竭19。其症状(如呼吸困难、乏力)非特异性,易被误诊为哮喘等常见病,平均诊断延迟达2-4年,确诊时多已进展至晚期910。PAH患者需长期接受多通路靶向药物联合治疗以延缓疾病进展,但复杂的用药方案(如多药联用)可能影响依从性211。目前治疗目标在于通过早期干预和动态风险评估,结合ERA、PDE5抑制剂及前列环素类药物等不同机制药物,改善患者生存质量并降低临床恶化风险。









 地 址: Rd.13 South, 31 km Ban Naphasuk,
SaithanyDistrict, Vientiane, Lao PDR
地 址: Rd.13 South, 31 km Ban Naphasuk,
SaithanyDistrict, Vientiane, Lao PDR  邮 箱: info@tlph.com.la
邮 箱: info@tlph.com.la  微 信: TLPH01; Whatsapp +856 20 76 814 389
微 信: TLPH01; Whatsapp +856 20 76 814 389  网 址: https: //tlph.com.la
网 址: https: //tlph.com.la